
OmniBiMol Variant Priority Pipeline
Lightweight, free-tier variant pathogenicity prediction models for the OmniBiMol bioinformatics platform.
What this does
Predicts whether a human missense variant is pathogenic or benign using:
- Precomputed genomic / conservation / protein scores (CADD, phyloP, phastCons, ESM-1b, NT, GPN-MSA, HyenaDNA)
- Biophysical protein features (BLOSUM62, Grantham, hydrophobicity, charge, volume, context)
- Gene-disease association evidence from OpenTargets clinical trials
Models included
| Model | Dataset | Features | AUROC | F1 | Size |
|---|---|---|---|---|---|
xgb_precomputed.json |
songlab/clinvar | 8 precomputed scores | 0.982 | 0.941 | ~391 KB |
rf_protein.pkl |
Rain021217/clinvar-pathogenicity | 20 protein features | 0.888 | 0.509 | ~19 MB |
lr_protein.pkl |
Rain021217/clinvar-pathogenicity | 20 protein features | 0.880 | 0.356 | ~1 KB |
Quick start
import pickle
import numpy as np
import xgboost as xgb
# Load XGBoost model (best AUROC)
model = xgb.XGBClassifier()
model.load_model("xgb_precomputed.json")
# Load preprocessing artifacts
with open("xgb_precomputed_imp.pkl", "rb") as f:
imputer = pickle.load(f)
with open("xgb_precomputed_scaler.pkl", "rb") as f:
scaler = pickle.load(f)
# Example input: [GPN-MSA, CADD, phyloP-100v, phyloP-241m, phastCons-100v, ESM-1b, NT, HyenaDNA]
# (obtain from VEP / precomputed tracks / songlab/clinvar)
sample = np.array([[1.5, 0.3, 0.7, -1.2, 0.1, -2.5, -0.8, 0.0]])
sample_imp = imputer.transform(sample)
sample_scaled = scaler.transform(sample_imp)
proba = model.predict_proba(sample_scaled)[0, 1]
print(f"Pathogenic probability: {proba:.3f}")
Batch inference
python inference.py --model xgb --input my_variants.csv --output scored_variants.csv
Gradio demo
python app.py
Data provenance
- songlab/clinvar — Precomputed missense variant scores from Cheng et al. (2023) Science (AlphaMissense), Notin et al. (2022) (Tranception), Frazer et al. (2021) Nature (ESM-1b), GPN-MSA, Nucleotide Transformer, HyenaDNA, CADD, phyloP, phastCons.
- Rain021217/clinvar-pathogenicity-prediction-dataset — Curated protein-level biophysical features derived from ClinVar missense variants with UniProt/VEP annotations.
- songlab/clinvar_vs_benign — ClinVar variant_summary filtered to missense variants with review_status and consequence annotations.
- opentargets/clinical_evidence — 2.7M clinical evidence records linking genes to diseases via clinical trials.
- gonzalobenegas/hgmd-omim-gnomad — Variants annotated with OMIM/HGMD traits for gene-disease grounding.
Extended experiments
Experiment 4: Gene-disease association scoring
Built from opentargets/clinical_evidence:
- 71,419 unique target-disease pairs scored
- Evidence score formula:
0.4*(max_phase/4) + 0.4*min(n_trials/10,1) + 0.2*min(n_datasources/5,1) - Ranges 0.04–1.00 (higher = stronger clinical evidence)
Experiment 5: Combined variant-to-therapy scoring
therapy_score = 0.5*pathogenicity_proba + 0.3*has_known_trait + 0.2*consequence_severity
Tier assignment:
- Tier 1 (Actionable): pathogenicity ≥ 0.9 + known disease association
- Tier 2 (High Risk): pathogenicity ≥ 0.7
- Tier 3 (Moderate): pathogenicity ≥ 0.5
- Tier 4 (Likely Benign): pathogenicity < 0.5
Files in this repository
| File | Description |
|---|---|
xgb_precomputed.json |
Best model: XGBoost on precomputed scores (AUROC 0.982) |
rf_protein.pkl / lr_protein.pkl |
Fallback models on protein features (no precompute needed) |
inference.py |
Production inference script (--model xgb or rf) |
app.py |
Gradio demo (single variant + batch CSV scoring) |
gene_disease_evidence.csv |
71,419 gene-disease pairs with evidence scores |
plots/*.png |
ROC, PR, confusion matrix, feature importance, distributions, precision@k |
metrics_summary.json / extended_metrics.json |
Full experiment results |
research_memo.md |
Literature review + integration recommendation |
Evaluation plots


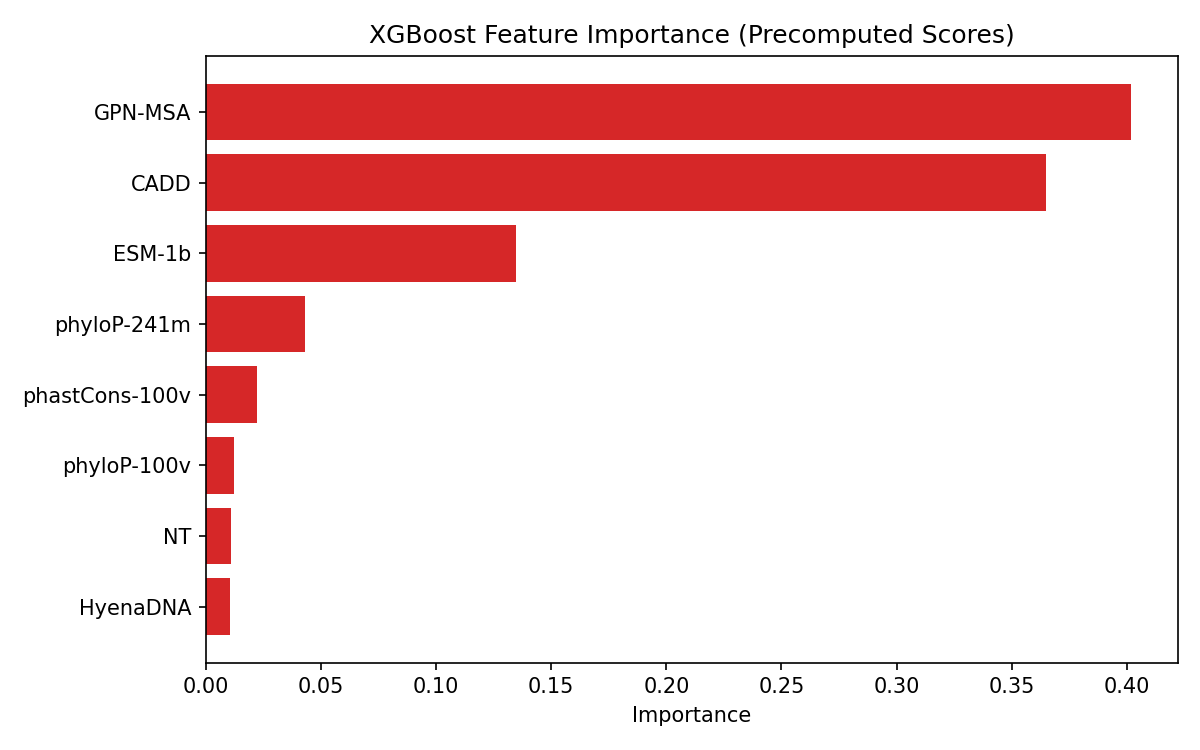
See plots/ for all 6 evaluation figures.
Training details
- Hardware: CPU-only (free-tier Hugging Face sandbox, 2 vCPU / 16 GB RAM)
- Framework: scikit-learn + XGBoost
- Train/test split: 80/20 stratified (exp2), provided train/test splits (exp3)
- Imputation: median
- Scaling: StandardScaler
- Class balancing:
class_weight="balanced"for RF/LR; dataset is naturally ~55:45 for exp2 - Total training time: < 5 minutes for all experiments
- Artifact size: ~19 MB total
Limitations
- Only missense variants are covered; frameshift, nonsense, splice are out of scope for the protein-feature models.
- The XGBoost model requires precomputed external scores (CADD, ESM-1b, etc.). If these are unavailable, fall back to the Random Forest on protein features.
- Models were trained on ClinVar 2023–2024 data; performance may drift on newer submissions.
- No clinical phenotype integration (OMIM, HPO) — this is planned for future OmniBiMol backend integration.
Integration into OmniBiMol
Patient VCF
│
▼
[VEP Annotation]
│
├─► Precomputed scores available? ──► XGBoost (AUROC 0.982)
│
└─► Only protein sequence? ──► Random Forest (AUROC 0.888)
│
▼
[Gene-Disease Lookup] ──► OpenTargets evidence score
│
▼
[Tier Assignment] ──► Tier 1/2/3/4
│
▼
[OmniBiMol Backend API] ──► Top-k + confidence + evidence
│
▼
[UI Report] ──► Sortable table → drug repurposing → wet-lab handoff
Citation
@misc{omnibimol_variant_priority,
title = {OmniBiMol Variant Priority Pipeline},
author = {OmniBiMol Team},
year = {2025},
howpublished = {\url{https://huggingface.co/omshrivastava/omnibimol-variant-priority}}
}
License
MIT
- Downloads last month
- -
Inference Providers NEW
This model isn't deployed by any Inference Provider. 🙋 Ask for provider support